REVIEW ARTICLE | https://doi.org/10.5005/jp-journals-10084-12140 |
Pitt-Hopkins Syndrome in Children
Department of Child Health Nursing, Kasturba Gandhi Nursing College, Puducherry, India
Corresponding Author: Sherin N Suria Prakash, Department of Child Health Nursing, Kasturba Gandhi Nursing College, Puducherry, India, Phone: + 91 9677025457, e-mail: sherinnithyaa@gmail.com
How to cite this article Suria Prakash SN. Pitt-Hopkins Syndrome in Children. Pon J Nurs 2020;13(4):93–95.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Pitt-Hopkins syndrome (PTHS) is the very rarest genetic mutation nervous system disorder. The children who are affected have classic facial features, high mental disability, global developmental delay, speech impairment, repetitive seizure complaints, and respiratory system abnormalities. In addition, the children have improper coordination (ataxia), repeated purposeless hand movements, sleep disorders, shortsightedness, and frequent constipation. Abnormalities in behavioral pattern are common, even though the children are social being with joyful disposition. Certain symptoms in some affected children resemble the symptoms of autism disorder. The unique clinical features of this condition are that the severity can vary from one child to another. The causative factor for this PTHS is changes in mutation at TCF4 gene. This mutation happens spontaneously in almost all the incidences, not because of the hereditary from family.
Keywords: Behavioral abnormalities, Classic facial features, Pitt-Hopkins syndrome.
INTRODUCTION
Children with Pitt-Hopkins syndrome (PTHS) have moderate to extreme mental handicap. The causative factor for this Pitt-Hopkins disorder is a transformation in the TCF4 gene. This mutation happens instantly, not as a result of family history. This condition was first depicted in the medication writing in 1992; but the genetic inclusion was found in the year 2008. Yet this disorder design was not clearly comprehended. The significant note is that the children who affected with Pitt-Hopkins disorder may not manifested all the symptoms explained below.1
Pitt-Hopkins syndrome is a condition characterized by mental disability, developmental delay, breathing impairment, recurrent seizures, and classic facial features.2
INCIDENCE
The occurrence of this disorder was precisely obscure. Presently around 500 people were recognized affected universally. Genetic and rare disease information center in the U.S. expressed that the affected children regularly run with misdiagnosed or undiscovered symptoms, making it hard to find the specific extent of the general population. This disorder influences both the genders and could influence the children from any racial foundation or ethnic populace.3
GENETIC MORPHOLOGICAL CHANGE
Pitt-Hopkins disorder is caused by an indistinct change in TCF4 gene. These genes give instructions for delivering proteins that do significant job in our body. Because of the mutation in the gene, the protein message becomes bogus, wasteful, missing, underdelivered, or overcreated. This protein creation significantly affects the elements of the frameworks of the body, including the brain. These proteins have significant part in the overall development of the children. It is extremely expressed by the development and affects the whole of the nervous system.
In case when the a mutation in TCF4 is defect causing, Pitt-Hopkins syndrome always happens in every new mutation, which means that almost every case the mutation has occurred in the time of the formation of the ova or sperm for that particular child only. And no one in their family is affected. This syndrome is usually not hereditary or carried by a healthy parent. If the transformation were to be carried from an affected individual to a kid, in most occurrences this could occur in an autosomal prevailing way. Genetic disorders are controlled by the compilation of genes for a particular traits happened due to the chromosomes from the parents. The abnormal gene is either acquired from parent or a new gene change in the affected individual. The risk of transmitting an abnormal gene from the affected parent to the kids is 50% for every pregnancy despite the gender of the subsequent child.
There are a few instances of the unaffected parent having more than one kid with this disorder. This is a very uncommon condition. Not all cells in the parent’s body undergo mutation, so the parent stay unaffected; however but the parent passes an alternate gene to his or her kids. As a result of these possibilities, it is estimated that the parents of the children with Pitt-Hopkins disorder can deliver around a 1–2% chance of having another affected kid, even if the parents are tested negative for gene change.4
CLINICAL MANIFESTATION
Infants with PTHS have very diminished muscle tone; this extreme hypotonia affects feeding in the children and diminishes the motor skills such as mobilizing. Difficulties in feeding occur in infant stage, but it resolves as the child grows up. The infant will be excessively quiet and sleepy. Some infants have microcephaly (Figs 1 and 2).5
Children with PTHS have an overall developmental delay, including head control. These children show extreme delay in learning even in holding and walking. Some children benefited out of knee and ankle brace to support and stabilize the weak and loose bony joints. When these kids attempt to walk, they demonstrate a wide stride and shaky way of walking. These children showing ataxia and delay in speech. Very few children attempt to utter few words; however, the vast majority of them are unfit to talk. A lot of them have superior repetitive language abilities; they could understand more information addressed to them. They can converse with themselves. These Pitt-Hopkins kids profited out of assistive communication gadgets which incorporate picture board and table-based talking gadgets. Many can learn to a degree with gesture-based communication.
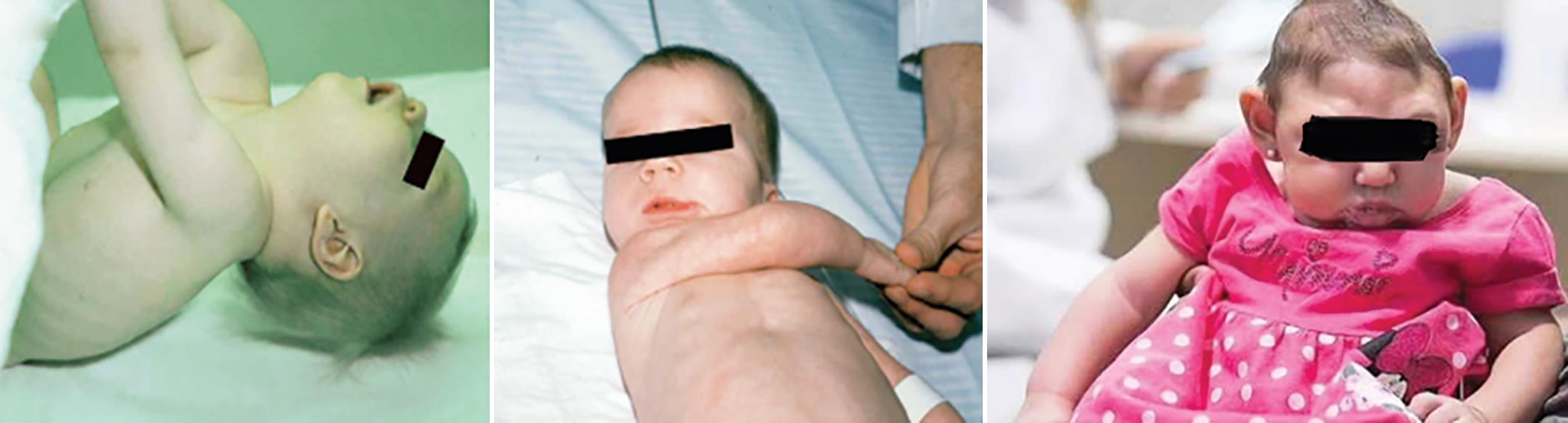
Fig. 1: Hypotonic and microcephalic features of infant (PTHS). https://www.mc.vanderbilt.edu/documents/neurology/files
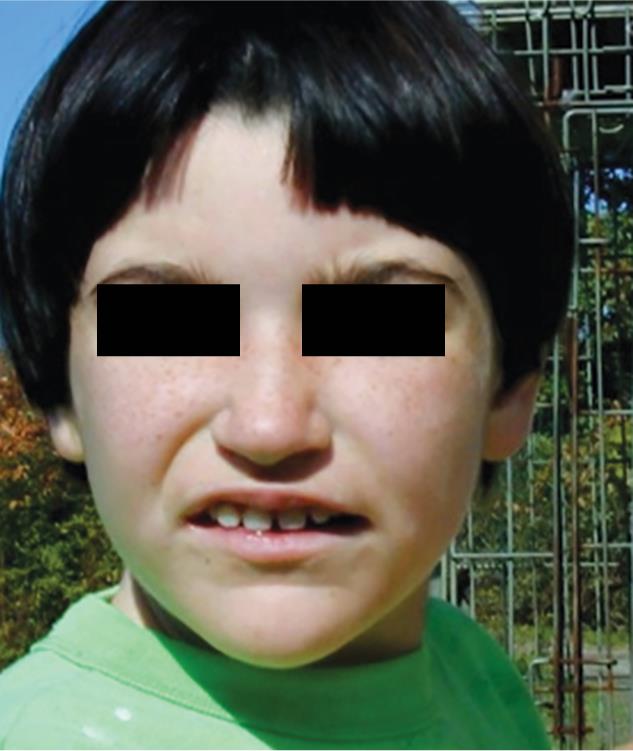
Fig. 2: Pitt-Hopkins syndrome of a 10-year-old child. https://www.nlm.nih.gov/files
Most of these kids have moderate to serious mental incapacity. However, speech and functional abnormalities make it difficult to decide the child’s true mental capacity. Technology advancements and newer therapies have enabled the affected kids to accomplish more what is really anticipated. The affected children have typical classic facial features. These features include an anomalous wide mouth with full lower lip, widely separated teeth, a board extension of the nose, a flared nostril, a sharp, downturned nasal tip, low set of ears with a cup-molded look, profound set of eyes inclined mildly upward with prominent supraorbital edge. These children have distended upper lip, that is, Cupid’s bow. Complete cheeks and some lower portion of the face and jaw might be prominent. These typical classic facial features become progressively noticeable at each developmental stage.6
The affected children can have impaired breathing pattern, i.e., recurrent sessions of irregular breathing and sometimes with hyperventilation and often apneic crises. Due to apnea, centralized bluish discoloration is observed in children. Strong emotions and exhaustion are the activating variables of sporadic breathing. The affected children are generally depicted as extremely jovial with a happy disposition nature, all the time chuckling and laughing. Giggling may happen instantly or at improper timings. A few kids are quiet and have self-absorption nature and need to confront trouble to be sociable. Frequent episodes of aggressiveness, hypershouting, and screaming are common in them while facing the unexpected circumstances.
These children are sometimes hyperactive, anxious, self-hurting, and shy. Children with Pitt-Hopkins disorder have repetitive hand moments, which may incorporate hand clapping, hand flapping, handwashing, thump sucking, head shakes, head banging, entire body shaking, teeth grinding, and hair pulling are also found. These children monotonously play with specific toys and may increasingly love with that toy. Fifty percent of these affected children endure with frequent episodes of seizure, which normally starts at the infant stage itself. Much of the affected children experience constipation and gastroesophageal reflux. Extra manifestations of this disorder incorporate excessive drooling, nearsightedness, strabismus, and astigmatism, and numerous cases scoliosis are also seen, roughly 33% of the affected male children have the complaints of cryptorchidism. Some minor anomalies seen include regular additional crease or missing on the thumb; and in some uncommon situation, some children are unable to twist their thumb because of missing ligaments. These children have redness of the skin underneath the nails. They also have superseding of toes. Most of the children have high pain threshold.7
DIFFERENTIAL DIAGNOSIS
Angelman Syndrome
It is an uncommon genetic and central nervous system disorder featured by serious developmental delay, learning disability, absence or near absence of speech, ataxia seen, outrageous jerky developments of the arms and legs experiencing happy disposition, and spontaneous laughing and smiling. Yet children with PTHS might be unable to speak but gradually learn to communicate through gestures and have enough receptive language ability. Angelman syndrome may have classical facial features but reflect the normal parent traits. This Angelman syndrome is caused by abnormal expression of UBE3A.
Mowat-Wilson Syndrome
Mowat-Wilson syndrome (MWS) is an uncommon genetic issue, which is obvious during birth or later in childhood. This issue is bound to have congenital gastrointestinal disorder known as Hirschsprung disease, where stenosis can be seen. Whereas in PTHS, Hirschsprung disease will not be present. Mowat-Wilson syndrome is caused by an abnormal mutation of ZEB2 gene.
Rett Syndrome
Rett syndrome is a progressive neurodevelopmental disorder. It solely affects the female children. In Rett’s disorder, individuals grow normally up to 7 months to 1½ years. After this point, they start to lose the past obtained abilities (developmental regression) and suffer from seizure disorder and microcephaly. Similar to PTHS, in this disorder the affected individuals develop hyperventilation. Rett syndrome is caused due to a change in MECP2 gene.8
CLINICAL INVESTIGATIONS
Pitt-Hopkins syndrome depends upon the complete child history, a through physical examination, and definite findings of the characteristics symptoms. There is an overlay of manifestations associated with PTHS and other similar neurological disorders. The absence of congenital disorders, which are structural and motor abnormalities present at birth, supports the diagnosis, which can be confirmed with molecular testing, indicating the change in TCF4 gene.9,10
TREATMENT MODALITIES
Treatment is directed toward the specific symptoms unique to each child and by the team of experts who will coordinated with medical geneticists or pediatricians. The members of the team consist of pediatric neurologist, gastroenterologist, ophthalmologist, speech pathologist, psychologist, psychiatrist, pulmonologist, pediatric nurse specialist, and psychiatric nurse specialist.11
Genetic counseling plays a significant role in the treatment process of the affected children and their family. Psychosocial support for the entire family is important as well. After the underlying affirmation of analysis, a developmental assessment has to be performed, and proper physical, occupational, speech, and feeding therapies are to be organized. Periodic reassessment and service modifications are to be taken consideration.
Severe speech impairment children to be taken into consideration at the earliest, and training should be provided with complementary and augmentative communication gadgets. Applied behavioral analysis will be useful to know the child’s strength and weakness to proceed with the treatment. A developmental pediatrician can help to sought out the behavioral problems and medication considerations for managing more aggressiveness in child.
Constipation is the most common issue in Pitt-Hopkins disorder among youngsters. The standard protocol is sufficient for treating constipation. In hyperventilation, issues or apneic attacks can be sought out by the instant drugs like acetazolamide. Seizure attacks may be controlled by anticonvulsants. In the vast majority of the children with PTHS, the ophthalmic glasses will help to resolve their shortsightedness; some of them need surgery for the ophthalmic problems. Periodical eye assessments are suggested.12
CONCLUSION
It is a genetic disorder, and there is no cure for the syndrome as of now. But with the proper management, the betterment of the child can be achieved. The best team of specialists can give a better life to the affected individuals at the maximum in their lifetime.
REFERENCES
1. Goodspeed K, Newsom C, Morris MA, Powell C, Evans P, Golla S. Pitt-Hopkins syndrome: a review of current literature, clinical approach, and 23-patient case series. J Child Neurol 2018;33(3):233–244. DOI: 10.1177/0883073817750490.
2. Bedeschi MF, Marangi G, Calvello MR, Ricciardi S, Leone FPC, Baccarin M, et al. Impairment of different protein domains causes variable clinical presentation within Pitt-Hopkins syndrome and suggests intragenic molecular syndromology of TCF4. Eur J Med Genet 2018;60(11):565–571. DOI: 10.1016/j.ejmg.2017.08.004.
3. De Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RCM. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis 2016;11(1):37. DOI: 10.1186/s13023-016-0422-2.
4. Rannals MD, Page SC, Campbell MN, Gallo RA, Mayfield B, Maher BJ. Neurodevelopmental models of transcription factor 4 deficiency converge on a common ion channel as a potential therapeutic target for Pitt-Hopkins syndrome. Rare Dis 2018;4(1): e1220468. DOI: 10.1080/21675511.2016.1220468.
5. Marangi G, Zollino M. Pitt-Hopkins syndrome and differential diagnosis: a molecular and development, cognition, and behaviour in Pitt-Hopkins syndrome. Dev Med Child Neurol 2012;54(10):925–931. DOI: 10.1111/j.1469-8749.2012.04339.x.
6. https://www.mc.vanderbilt.edu/documents/neurology/files.
7. https://www.nlm.nih.gov/files.
8. Peippo M, Ignatius J. Pitt-Hopkins syndrome. Mol Syndromol 2012;2(3–5):171–180. DOI: 10.1159/000335287.
9. Whalen S, Héron D, Gaillon T, Moldovan O, Rossi M, Devillard F, et al. Novel comprehensive diagnostic strategy in Pitt-Hopkins syndrome: clinical score and further delineation of the TCF4 mutational spectrum. Hum Mutat 2012;33(1):64–72. DOI: 10.1002/humu.21639.
10. https://www.nature.com/articles/gim2009121.
11. https://rarediseases.info.nih.gov/diseases/4372/pitt-hopkins-syndrome.
12. https://ghr.nlm.nih.gov/condition/pitt-hopkins-syndrome.
________________________
© The Author(s). 2020 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.